Introduction
The replacement of synthetic antioxidants by “safer natural mixtures” has been increasingly advocated, nowadays by food industry (Kiokias and Varzakas 2014). This trend has been imposed by the worldwide preference of consumers for the use of natural antioxidants, some of which may exist inherently in foods or be added intentionally during their processing (Pantavos et al., 2015; Kiokias et al; 2009b). Among these, carotenoids comprise the group of the most abundant micronutrients in vegetables and fruits, that impact human nutrition contribute to fragrance and flavor of various products (Shumskaya and Wurtzel, 2013, Liu et al., 2013). Decades of research on carotenoids has improved our understanding of the role of these ubiquitous pigments, which have emerged as important players exerting a protective role against diseases associated with aging, including cancer, cardiovascular disease, cataracts, and age-related macular degeneration (Bowen et al., 2015; Bermudez et al., 2005;). Though the in vivo antioxidant activity of dietary carotenoids (e.g. beta-carotene, lycopene) is well established (Kiokias, et al., 2008; Dimakou and Oreopoulou 2012), there is still controversial scientific evidence about the factors that influence their in vitro properties in oil-based systems (Meléndez-Martínez et al., 2014; Kovary et al., 2001).
Carotenoids are synthesized in nature by plants and many microorganisms (Rodriguez and Stange, 2013; Bai et al., 2015). Most carotenoids are 40-carbon terpenoids having isoprene as their basic structural unit A general subdivision is into: (i) “carotenes” which are strictly hydrocarbons (a– and b-carotene, lycopene) and (ii) “xanthophylls” (lutein, bixin, capsanthin etc), which contain polar ends groups reflecting an oxidative step in their formation (Bohm et al., 1999; Faure et al., 1999).
Natural dispersions of carotenoids are used in coloring fat based foods such as margarine, butter, shortenings, cheese, and french dressings (Kiokias and Gordon 2004). Water-dispersible forms of carotenoids have been developed for the coloring of water-based foods such as orange-type beverages, cake mixes, puddings, dried and canned soups (Boileau et al., 1999).
A certain body of literature has focused on functional properties of natural extracts of carotenoids such as paprika, lycopene, lutein-rich extracts (Kiokias & Gordon, 2003b,a; Kiokias & Oreopoulou 2006, Kiokias et al., 2009). However, analysis of these preparations for the separation and identification of the carotenoid pigments is considered as essential in order for researcherS to explain and further optimise the functional properties of the carotenoid plant extracts.
Classical method of analysis of carotenoid pigments using column chromatography, paper chromatography and TLC potentiate isomerisations and transformations during the separation (Kiokias 2002). Reversed Phase High Performance Liquid Chromatography (HPLC) is the analytical method of choice for separation, quantification and structural characterization of the naturally occurring and synthetic carotenoids as well as for their most important metabolic products (Rodriguez-Amaya, 2015). Additionally, the introduction of the photodiode array detector has facilitated the identification of carotenoids after HPLC separation by using spectral characteristics (Shoefs et al., 1995).
This research work first focused on the separation of carotenoids (via the development of a suitable saponification process) from a range of commercially available extracts of natural origin, such as extract of palm oil carotenes, marigold extract, annatto (water and lipid soluble preparations) tomato extract, and paprika extract. In other studies these commercial preparation of natural origin have been examined for investigation of antioxidant activities in vitro and in vivo, therefore a better understanding of their active carotenoid content would be decisive for further developing and optimising their future application as food or dietary supplements.
Material and Methods
Materials
For this experimental work, the following carotenoid standards have been used: all trans synthetic b-carotene (purified 95%), and trans-apo-b-carotenal (20%) suspended in vegetable oil, xanthophylls and lycopene standards (provided by Sigma CO, Poole, UK); capsanthin (94%) and capsorubin (98%) provided by Carotene Nature GmbH (Switzerland). Furthermore, the following carotenoid extracts were donated by Overseal Foods Ltd (Swadlincote, UK) for analysis in the present study: (i) Natural extract of palm oil carotenes; (ii) Natural tomato preparation extracted from ripe tomatoes; (iii) Marigold flower (Tagetes erecta) extract rich lutein esters (iv) Paprika oleoresin, extract of paprika (Capsicum annuum) soluble in vegetable oil (v) Extracts of natural annatto (Bixa orellana) suspended in refined vegetable oil and water soluble norbixin powder respectively.
Methods for carotenoid analysis
Saponification prior to HPLC Analysis
Carotenoid extracts were saponified prior to their HPLC Analysis by a modification of a method proposed by Hart & Scott (1995). More specifially, the natural extract (10 mg), diluted in 10 ml of the mobile phase, was saponified with an equal volume of 10 % potassium hydroxide in methanol (under nitrogen in the dark with stirring) for 1h at room temperature. The carotenoids were extracted from the KOH/ methanolic phase by careful shaking with 20 ml light petroleum (containing 0.1 % BHT), and 20 ml 10% sodium chloride in a separating funnel. The lower KOH/MeOH/aqueous phase was removed to another separating funnel and extracted 2 more times with 20 ml of light petroleum. The light petroleum phases were combined in a separating funnel and washed with water until the washings were neutral to pH paper and transferred to a 100 ml round bottom flask from where solvent was evaporated on a rotary evaporator at 35°C. The residue was redissolved by ultrasonic agitation in 10 ml of the mobile phase and diluted with the mobile phase to a suitable concentration and filtered through a 0.4 mm syringe filter.
HPLC Analysis for identification of carotenoid pigments
A Hewlett Packard 1050 liquid chromatograph, equipped with a diode array detector, was used for the analysis of the various carotenoids, with data collected by a Chemstation 7. Sample (20 ml) was injected onto a Spherisorb ODS-1 reversed phase column (250 ´ 4.6 mm i.d., 5 mm particle size), protected by a guard column (ODS-1). Following a series of experiments, the specific conditions were optimized and used for the various carotenoid extracts.
(A) Analysis of extracts rich in more lipophylic carotenes:
For the HPLC analysis of palm oil-carotenes and tomato extracts and isocratic elution of the following solvent systems were used:
Solvent-1: Acetonitrile (MeCN)/methanol(MeOH)/ tetrahydrofuran (THF)/
/ammonium acetate (1% w/v) in the ratio 68:22:7:3 (%). Flow rate : 1.0 ml/min
Solvent-2: (MeCN)/(MeOH)/(DCM) in the ratio 75:20:5 Flow rate : 2.0 ml/min
The mobile phase contained 0.1% butylated hydroxytoluene (BHT) and 0.05 % triethylamine (TEA), whereas methanol used for the mobile phases contained 0.05 mM amonium acetate.
Detection at: 450 nm for caroteneS, 472 nm for tomato extract.
(B) Analysis of extracts rich in more hydrophilic xanthophylls:
For the analysis of paprika extract the following two solvent systems were used and compared
Solvent-3: Gradient elution of (I) 75:25 ( v/v) acetone-water, (II) 75:25 (v/v) acetone-methanol. Gradient elution : 0 % (I) to 65 % (II) in 10 min
to 80% (II) B in 30 min
to 20% (II) B in 60 min
Flow rate : 1.5 ml/min. Detection at 450 nm.
Solvent-4: Isocratic elution of Acetonitirile/ 2-propanol/water in the ratio 39:57:4. Flow rate: 1.0 ml/min. Detection at 450 nm.
Solvent-5:For the analysis of marigold flower extract,
Gradient elution: methanol – ethyl acetate 60-40
to methanol- ethyl acetate40-60
Linear gradient over 20 min. Flow rate: 2 ml/min , detection at 446 nm.
Solvent-5
For the analysis of water soluble annatto preparation (rich in nobixin) an isocratic of acetonitrile: aqueous acetic acid (0.4% v/v ). Flow rate: 1 ml/mn, detection at 436 nm. For the oil soluble annatto, rich in bixin, the solvent-1 (described above for analysis of carotenes) was used with detection at 450 nm. Carotenoid samples were examined for the presence of tocopherols by a normal phase HPLC analysis as described by the AOCS official method Ce 8-89 (AOCS, 1989).
UV Spectroscopy for the determination of total carotenoid content
Following experimentation and literature searching for the selection of the appropriate solvent, (as decsribed in table-2) solutions of the various carotenoid extracts (1mg/100 ml) were prepared, and a spectrum of each one (200-500 nm) was obtained. The specific absorbance, at the lmax of the major carotenoid for each extract was determined with a UV-Vis Spectrometer (Lambda Bio 20, Perkin Elmer, Basingstoke, UK). Absorbance values (n=6) compared with absorptivity values (extinction coefficient of E1 cm,1% ), reported in the literature (Kiokias, 2002; Cabrera, et al., 2010), and offered the basis for the approximate determination of the total carotenoid content of the natural extracts.
General aspects of HPLC carotenoid analysis in plant extracts
Carotenoid extracts from natural sources are usually saponified to remove chlorophylls and unwanted lipids and to hydrolyse carotene esters (Khachic et al., 1997). Organic solvents extract carotenoids, whereas fatty acids that precipitate as soaps and glycerol are retained in the aqueous phase.
However some workers have observed a significant loss of carotenoids after saponification. Miki et al. (1990) showed that saponification was accompanied by significant loss of xanthophylls, particularly the epoxycarotenoids, whilst losses of carotenes were not significant. Daood et al (1996) suggested that loss of xanthophylls during saponification can be effectively reduced by using antioxidants such as butylated hydroxy-toluene (BHT).
Most current HPLC separations of carotenoids are performed by using short (<20 min) isocratic elution with commercially prepared Reversed Phase (R.P) Columns. In these columns, silica particles are chemically bonded with a non-polar stationary phase, which is less polar than the solvent of the mobile phase (Ball, 1988). Therefore the polar compounds (such as xanthophylls) elute first while the non-polar carotenes are retarded more by the hydrophobic stationary phase and elute later. Unlike adsorption (normal phase) HPLC, reverse phase systems are capable of separating positional isomers of carotenoids, such as a- and b-carotene, and there is a little risk of on-column carotenoid degradation on the non polar bonded phase (Saleh & Tan, 1991). In addition Reverse Phase packings are not sensitive to moisture content, whereas normal phase packings are, and they re-equilibrate rapidly following gradient elution.
The most common column packing used for carotenoid separation is octadecylsilane (ODS), which is a highly retentive stationary phase (Craft, 1992). This packing material was used in this project (Zorbax ODS), to avoid the problems of poor sample solubility associated with partly aqueous mobile phases.
In the present study and, a qualitative estimation of the individual identified carotenoids in their mixtures (following the HPLC analysis) was given by the following ratio: % PA = (peak area of each carotenoid) / total peak area of all the eluted pigments in the chromatogram.
HPLC analysis of natural extracts (palm oil, tomato) rich in carotenes
Palm oil carotenes
Carrot extracts, carrot oil, and palm oil related extracts are available in the market and their main components are a- and b-carotenes (Kiokias and Gordon, 2003a). Purified crystalline products, dispersions of microcrystals in oil and carrot oil are also commercially produced.
In the initial HPLC experiments, a natural extract of carotenes from palm oil was analysed. Prior to the HPLC analysis, a saponification procedure was applied, in order to remove unwanted lipids and other pigments that could interfere with the carotenoids.
Lietz and Henry (1997) has reported losses of carotenoids during HPLC analysis of red palm oil after the saponification of the extract. This issue has been examined in the frame of the present study for the tested palm oil extracts. Very little loss of carotenes (<4%) was estimated in the saponified sample, following measurements of absorbance at 450 nm (monitoring decrease of Abs450 of 1 mg/ml solution of carotene extract in THF).
A mobile phase of acetonitrile/THF/methanol/ammonium acetate (solvent-1) was used (as described in section 2.2), which has been generally found to be the most appropriate for the separation of carotenoids from natural extracts (Hart & Scott, 1995) for reasons that are given below. In selecting the mobile phase for carotene analysis it must be remembered that carotenes are practically insoluble in water, so this should be avoided or only be incorporated sparingly as a solvent modifier. The primary solvent should be a weak organic acid with low viscosity, thereby allowing use with a range of modifiers, yielding adequate carotenoid solubility, and low backpressure. These criteria are met satisfactorily by acetonitrile and methanol. Generally acetonitrile has been used because of its lower viscosity and slightly improved selectivity when using monomeric C18 columns. Most frequently, strong solvents such as tetrahydrofuran (THF) or ethyl acetate have been used as modifiers for carotenoids separation (Zakaria et al., 1989). A similar solvent system has been used by Scott and Eldridge (2005) in the carotenoid analysis of fresh, frozen and canned foods.
A solvent modifier, also reported in the analysis of carotenes, is ammonium thiocyanate, which is proposed to reduce the on-column degradation of carotenoids, in particular when small quantities of carotenoids are analysed (Handelman et al., 1992). In general, the exact action of these solvent modifiers is not completely clear, but it is suggested that the improvement in recovery, is caused by buffering of the acidity in the mobile phase, or more likely the acidity of the free silanols in the stationary phase or by preventing reactions with free metal ions.
As shown in the table-1, the palm oil extract contained mainly a- and b-carotenes while lycopene was also detected at very low levels. This specific mobile phase is very selective not only in distinguishing a-carotene from b-carotene, but also in separating various isomers of b-carotene, such as all trans (which is the predominant), and 9-cis b-carotene. Table-1, is listing the retention times as well as the relative peak areas (% PA) of the eluted carotenoid pigments following the analysis of their saponified samples.
According to the results, all trans b-carotene (Rt=11.2 min, PA=54.5 %) was the major carotenoid, followed by cis-b-carotene (Rt=11.8 min, PA=23.5 %) and a-carotene (Rt=10.7 min, PA=19.1 %), whereas lycopene (Rt=7.8 min, PA=2.9 %) was also present in traces. However, it must be stressed that isomerisation of carotenes was observed in the present study as a result of saponification. A significant increase (33%) of all-trans b-carotene was accompanied by losses of cis β-carotenes (42%) and a-carotene (35%), when peak areas of these compounds between saponified and unsaponified samples were compared. Actually, structural transformation of some carotenoids has been reported in literature, as a consequence of the saponification prosedure (Daood et al., 1996). Aebisher et al (1999) by using similar HPLC conditions to those used in the present study, found the same order of elution of various carotene isomers, but the retention times were a few minutes longer.
In the present study, a mobile phase of acetonitrile/methanol/dichloromethane (MeCN:MeOH:DCM): 75:20:5 (solvent-2) was also used. Triethylamine (TEA 0.1%) was added to the mobile phase to increase the recovery of carotenoids ammonium thiocyanate (0.05 M in the methanol of the mobile phase), was also used as a solvent modifier.
In comparison with HPLC performed with acetonitrile/THF/methanol/ammonium acetate (solvent-1), it was found that by using solvent-2:
(a) α-carotene was eluted ~3 min earlier than β-carotene, but it was not possible in the obtained chromatograms to distinguish between all-trans from cis-β–carotene.
α- and β-carotene overall were eluted later ((Rt=32.5 and 35.5 min, respectively).
Therefore, it was concluded that solvent 1, offers a more selective system for analysis of the carotene extracts.
For the quantification of b-carotene in the natural extract, the external standard method was used. This technique avoids the problem of finding a suitable internal standard. Following this method, a series of standard b-carotene solutions in the range of 20-100 ppm were prepared. Quantification and quality control of carotenoids with external standards calibration method by use of HPLC chromatograms peak areas has been reported to work very well by Scott and Eldridge (2005) in the analysis of various food items.
In the present study, equal amounts of each standard were chromatographed and the following calibration curve of peak area (PA) against concentration (ppm) was prepared:
C (ppm)= 18*10-3 PA r2=0.98 (n=6)
According to the calculations (and considering that a-carotene by having a similar molar absorptivity with b-carotene can be also quantified with the same equation), it was found that a saponified carotene extract of 20 ppm concentration contains, 12.4 (±0.4) ppm of all trans β-carotene (~62%), 4.1 (±0.3) ppm of 9-cis β–carotene (20.5%) and 3.5 (±0.2) ppm of a-carotene (~17.5%).
Tomato extract
The next sample for HPLC analysis was the tomato extract, which according to the product specifications have lycopene as the major carotenoids pigment. Lycopene is a tetraterpenic C40 carotenoid that absorbs light in the red region being thereby responsible for the color of tomato and watermelon (Gann et al, 2012).
For industrial uses natural lycopene is readily purified from tomato processing wastes (Silva et al., 2013).
According to Hakal & Heinonen (1994), a solvent mixture of acetonitrile/methanol/THF is appropriate as a mobile phase for carotenoid separation in a natural extract of lycopene, when a Zorbax ODS column was used.
In the current study, the tomato extract was analysed by the use of the same mobile phase (solvent-1) that was previously described for the analysis of palm oil carotenes. Similarly, a quantity of the extract was saponified with the method described in section 2.1 and the final residue was made with the mobile phase to a final volume of ~100 ppm.
From the relative peak areas (table-1) it was deduced that lycopene was the major component having a peak area of ~87%, (Rt=7.8 min). In addition, a significant proportion of all trans b-carotene (~11.5%, Rt=11.3 min) was present in the tomato extract along with traces of a-carotene (~1.5%, Rt=10.7 min), and cis–β-carotene (Rt=11.9 min, PA=0.9 %). In fact, the same components were identified by Hakall & Heinonen (1994) with consistent similar and retention times, whereas additionally found hydroxycarotenoids (lutein or zeaxanthin) that were not identified in the sample analysed in the present study.
HPLC Analysis of natural extracts (marigold, paprika, annatto) rich in xanthophylls
Paprika extract
Extracts of paprika (Capsicum annuum), rich in capsanthin carotenoids, are the oldest and most important natural carotenoid food colours, used as dry powder or the oil extract of coloring and flavoring components from the pods known as oleoresin of paprika (Giuffrida et al., 2013; Park, S-Y et al., 2014). Oleoresin of paprika, unlike ground paprika, is a liquid product completely soluble in oils and therefore does not impart specks or vegetable tissues to the food product (Pérez-Gálvez and Mínguez-Mosquera, 2004). A great advantage of the oleoresin is the possibility of standardizing the color of the oil extract since the paprika depends on various factors such as drying temperature, moisture content and storage conditions. Topuz et al. (2011) have recently examined the influence of different drying methods on carotenoids and capsaicinoids of paprika.
Because of the complexity of the carotenoid mixtures in paprika extracts, various mobile phases have been described for the isolation and quantification of the carotenoids present. Several authors have reported that both qualitative and quantitative analysis of carotenoids in natural paprika extracts can be achieved without prior saponification of the sample (Deli et al., 1992). According to Daood et al. (1996), it is important to avoid alkaline hydrolysis of carotenoid esters present in paprika extracts, because this step often leads to artefact formation from the pigments giving rise to a greater experimental error.
In the present study, samples of the paprika extract were analysed both with and without saponification to study the effect of this process. As described in section 2.2 for the identification of paprika extract pigments, two HPLC mobile phases were tested in this study. Initially, an isocratic elution of an acetonitirile based mobile phase was used (solvent-3). Saponification helped the identification of the pigments, in the paprika extract (figure-1). In the sample, which had not be saponified, the carotenoid monoesters are distinguistable from the diesters but full identification cannot be achieved. After saponification which caused the hydrolysis of paprika esters and the removal of unwanted lipids and interfering pigments, the carotenoid pigments were clearly eluted as: capsorubin (Rt=6.5 min, PA= 35.4%), capsanthin (Rt=7.5 min, PA= 55.2%), b-carotene (Rr=10.5 min, PA=9.4%). The identification of pigments was based on the use of pure carotenoid standards. The relative amounts of carotenoids observed here are similar to values reported in the literature for saponified extracts of paprika (Biacs et al., 1989; Deli and Molnar, 2002). The results are consistent with the specification of the sample, which stated that capsanthin and capsorubin are the major pigments in the paprika extract.
 |
Figure 1: Effect of saponification on HPLC Analysis of Paprika extract Click here to View figure |
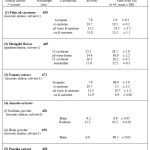 |
Table 1: Summary of the results of Reverse Phase HPLC Analysis of natural carotenoid extracts (by use of the solvent systems described in sections 2.1) Click here to View table |
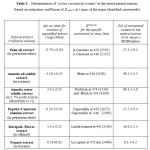 |
Table 2: Determination of “active carotenoid content” in the tested natural extracts (based on extinction coefficient of E1 cm,1% at λ-max of the major identified carotenoids) Click here to View table |
As an alternative system, a gradient elution with a mixture of acetone-water and methanol was also used for the analysis of the saponified paprika extract (solvent-4). Identification of peaks was based on the chromatograms reported by the same authors. Similar to what earlier observed with isocratic elution, the main identified peaks were capsorubin (Rt=6.1 min, PA=39.5%) and capsanthin (Rt=9.0 min, PA=52.1%), with traces of b-carotene (Rr=12.5 min, PA=8.4%).
Comparing the 2 HPLC solvent systems for the analysis of parika’s saponified sample, it can be concluded that by using the isocratic acetonitirile-based mobile phase we had a slightly earlier elution of the identified carotenoids (shorter retention times). However no significant differences were reported concerning the identified pigments and their proportions in the analysed carotenoid mixtures as expressed by their % PA of the obtained chromatograms.
Analysis of Marigold extract
The next analysed sample of xanthophylls was a refined extract of marigold flower (Targeta Erecta). Marigold extract is a commercially available food colorant (mainly of yellow color), which is considered as a stable source of the dietary carotenoid lutein, a normal constituent of human plasma and retina (Vargas and Lopez, 1997, Xiao-Dan Fan et al., 2015).
For the analysis of the marigold extract, the HPLC system was modified compared to the one used for the carotenes. Gradient elution is generally preferred for the separation of xanthophylls from natural extracts as they have a wider range of carotenoid polarity (Šivel et al., 2014). Following a series of experiments, a gradient elution of a solvent mixture consisting of methanol and ethyl acetate was selected which as the most appropriate for the analysis of the marigold flower extract.
As with the rest of the analysed extracts, the marigold sample was saponified before HPLC analysis. According to the product specifications, this extract contains mainly free lutein and lutein esters (mainly lutein dipalmitate) and small quantities of related carotenoid esters (including zeaxanthin). Similarly to what previously reported for paprika extract, there was a positive effect of the saponification on the identification of carotenoid pigments. In the chromatogram of the unsaponified sample, a larger number of peaks are present some of which represent lutein isomers, others may correspond to other pigments (such as chlorophylls) that exist in the sample, making thereby very challenging the separation of the carotenoid pigments.
Therefore for the subsequent quantification of the main carotenoid pigments in the tested marigold extract, the saponified sample was further elaborated. Indeed, the gradient elusion revealed four distinct peaks corresponding to different lutein isomers. Firstly, 13 cis-lutein eluted (Rt=12.1 min, PA=28.5%) followed by all-trans lutein, which was the major component (Rt=14.6 min, PA=38.8%). In addition, 9-cis lutein (Rt=16.1, PA=20.2%) and zeaxanthin (Rt= 17.6, PA=12.5%) were present in lower amounts.
Retention times and components were similar to those found by Vargas & Lopez (1996) in marigold flower extracts. All the lutein isomers eluted close to each other (in a range of 5 min), and no carotenes were detected in the samples. Hadden et al (1999) had identified the same carotenoids by using normal phase HPLC systems, although also reported small quantities of lutein oxidation products in the saponified sample.
The compositional determination of carotenoids in the marigold extract, by revealing almost ~90% of lutein isomers in the tested saponified sample, further to its potential as food colorant it is also confirming its dietary value, and could also offer the basis for future applications in nutritional supplements.
Analysis of annatto extracts
The term annatto includes a series of coloring preparations consisting of carotenoid-type pigments; all based on extracts of the seed of the tree Bixa orellana which grows abundantly in the tropics (Chisté et al., 2011; Viuda-Martos et al., 2012). Bixin is the main component of oil-soluble preparations, and norbixin of the water-soluble products. Norbixin powder is obtained from fresh annatto seeds by means of alkaline extraction following by a precipitation of the colorants with a mineral acid. The extract is then filtered pressed and dried at controlled temperature in order to permit its preservation (Scotter et al., 1998). It produces orange solutions, suitable to color foods including cheese products, butter, margarine and salad dressing. The major coloring component of annatto is the apo-carotenoid 9-cis-bixin, the solubility of which is commercially achieved by heating a preparation of the seeds in oil to a maximum temperature of 130˚C in vacuo. Under these conditions, 9 cis bixin undergoes isomerisation to produce oil-solutions containing variable proportions of the pigment dependent on extraction temperature and time (Scotter, 2009).
In the present study, the tested water-soluble and lipid-soluble annatto extracts were in powder form with high degree of purity as also evidenced by UV-vis spectroscopy Furthermore, since these were not natural dispersions in oil (e.g. as in case of palm oil, paprika and tomato extracts) no need for saponification prior to by HPLC analysis was identified. Therefore, HPLC Analysis with isocratic elution of acetonitrile based solvents was used to assess the purity of the annatto extracts. The water-soluble preparation (norbixin powder) was analysed using an isocratic mobile phase consisting of 65:35 acetonitrile: aqueous acetic acid (0.4%) (solvent-6). Norbixin as expected was the main pigment (PA=93.2 %, Rt=11.2 min), whereas bixin (lipid homologue of norbixin) eluted a bit earlier (PA=6.8%, Rt= 9.2 min) was also present as a minor component. Extensive HPLC analysis of norbixin has been carried out by Scotter et al.(1996) who identified all-trans and di-cis isomers of norbixin in annatto extracts with a reverse-phase HPLC system, utilising a photodiode-array detector. For the analysis of bixin powder the same mobile phase (MeCN/MeOH/DCM) and HPLC conditions to what used for carotene analysis was applied (solvent-1). It was proven quite efficient in achieving an early and clear elution of the bixin peak (Rt=6.8 min) that was the only carotenoid pigment detectable in this annatto powder (table-1).
Determination of tocopherols in carotenoid extracts
The determination of tocopherols, if present, in the oil dispersions of natural carotenoid extracts is deemed essential and potentiually critical in order to explain their functional properties. For instance, when the natural extracts are tested for antioxidant activity in bulk oil or emulsion systems (Kiokias et al., 2009a) it is important to know their tocopherol content given that tocopherol-carotenoid combination may under certain conditions account for synergistic activity (Kiokias and Oreopoulou, 2006). In these experiments, an HPLC system with fluorescence detection was used to analyse the tocopherols in all the carotenoid extracts (as discussed in section 2.2). Initially, standard curves of various tocopherols (concentration vs peak area was prepared) by HPLC analysis. As a type of example, for a-tocopherol the quantification was made by relating concentration and peak area of the chromatograms via the following equation:
PA= 4.5 x C (ppm) + 6.3 r2=0.98
According to the results, annatto extracts (powder forms with high carfotenoid purity) were devoid of any tocopherols. Furthermore, the normal phase HPLC analysis of marigold, paprika and tomato extracts revealed that these contained only a-tocopherol (no other tocopherol isomers were identified). More specifically, a-tocopherol was present in traces in the marigold extract (at ~1.5%) in small proportion in the paprika extract (at ~5% ) and at quite significant concentration in the tomato extract (at 11%). In particular for the tomato extract, the high concentration of a-tocopherol should be taken into careful consideration when investigating for any antioxidant activity of this specific preparation following its addition in model food systems.
Determination of “active carotenoid content” in natural extracts
In the last phase of this experimental work, it was considered useful to produce estimations of “active carotenoid concentrations” of the tested natural extract by use of visible absorption spectroscopy. This term was selected in order to reflect the concentration of the main identified carotenoid pigments in each tested preparation. Obviously, it concerns approximate estimations which would be more helpful to understand and explain the functional properties of these extract as well as to adjust their concentrations in experiments where they are added in food systems. Historically, quantification of carotenoids in natural extracts has mainly been achieved by measuring total absorption at a specific wavelength (Hart & Scott, 1995).
Carotenoids by virtue of their various colors exhibit very strong absorption in the visible region of the electromagnetic spectrum (Brubacher et al.,1985). In any given solvent λmax values increase as the length of the chromophore increases. Carbonyl groups, in conjugation with the chromophore system extend the chromophore, whereas other substituents, such as hydroxy or methoxy groups do not have any effect. For example, β-carotene and its hydroxy derivatives β-cryptoxanthin, and lutein have identical spectra with λmax at around 450 nm but echinenoene and canthaxanthin (ring keto-group), have their λmax at 461 and 478 nm respectively. Visible spectroscopy was applied to determine the total content of the specific carotenoids in natural extracts. Although, saponification was useful in separating and identifying the carotenoid pigments prior to HPLC analysis (as discussed in section 3.2). However, this process was not applied in the samples of the natural extracts that were used of this spectrophotometric analysis. This was chosen in order to estimate the “active carotenoid concentration” in the real commercially available extracts which are added as such for coloring or functional applications in food systems (and without any further chemical processing such as saponification). Following experimentation and literature searching for the selection of the appropriate solvent, solutions of the various carotenoid extracts (1mg/100 ml) were prepared, and a spectrum of each one (200-500 nm) was obtained. The specific absorbance, at the λmax of the major carotenoid for each extract was calculated, and compared with absorptivity values (extinction coefficient of E1 cm,1% ) reported in the literature (Kiokias, 2002; Cabrera, et al., 2010).
According to the results, (table-2) the “active carotenoid concentrations” were as following:
- ~56 % for marigold extract (mainly lutein as esters),
- ~26 for the palm oil extract (mainy as a-, b-carotenes),
- ~6% for the tomato extract (mainly as lycopene, with traces of carotenes),
- ~16% for the paprika oleoresin extract (mainly expressed as the total of esters of capsanthin and capsorubin),
- ~100% for the oil soluble annatto powder (expressed as bixin)
- ~ 96% for the water soluble annatto powder (expressed mainly norbixin, with traces of bixin).
These results could be applied in the case that we would like to compare the functional properties of all these natural extracts, for instance, their antioxidant potential against the autoxidation of an oil-in-water emulsions. Assuming that, for example, we want to investigate the effect of addition of each extract at a concentration of 1 mg/kg of active carotenoid content, (having as basis the almost pure -in bixin- annatto extract) the following quantities of each extract should be diluted in 1 krg of each emulsion treatment (keeping one without carotenoids as a control): 1 mg of oil-soluble annatto powder, 1.04 mg of water soluble annatto, 1.78 mg of marigold extract, 3.84 mg of palm oil carotenes extract, 6.26 mg of paprika extract, 16.7 mg of tomato extract.
Therefore the results of this work can offer the basis for the design of further experiments the potential functional properties of these natural extract following their in vitro additions in food model systems.
Conclusions
The following conclusions can be drawn from this experimental work:
- Saponification prior to HPLC analysis of natural oil dispersions (paprika, marigold) resulted in the removal of unwanted pigments and lipids and the hydrolisation of esters, therefore facilitated the identification of major carotenoid pigments in the extracts. In the analysis of palm oil carotenes, though, resulted on isomerization with losses (at ~40%) of cis b-carotene and a-carotene.
- Reverse phase HPLC systems are capable of separating positional isomers of carotenoids with isocratic elution of the solvent systems being more efficient for the separation of carotenes or pure annatto preparations and gradient elution overal more selective for the analysis of xanthophylls.
- Determination of the active carotenoid content in the natural extracts could be useful for further standrdising and optimising their future applications both as food additives and nutritional supplements
References
- Aebisher, C.P., Shirley, J., & Shuerp, W. (1999). Simultaneous determination of retinol, tocopherols, carotenes and xanthophylls in plasma by RP HPLC. Methods in Enzymology, 299, 351-356.
- AOCS (1989). Official methods and recommended practices. D.Firestone. Illinois, AOCS Press.
- Bai, C., Capell, T., Berman, J., Medina, V., Sandmann, G., Christou, P., and Zhu, C (2015). Bottlenecks in carotenoid biosynthesis and accumulation in rice endosperm are influenced by the precursor–product balance. Plant Biotechnology Journal, (published online, DOI: 10.1111/pbi.12373).
- Ball, G.F. (1988). Fat soluble vitamin assays in food analysis-a comprehensive review. Elsevier Applied Science. New York.
- Bermudez, I.O., Ribaya-Mercado, D.J., Talegawkar, A.S., Tucker, L.K. (2005) Hispanic and Non-Hispanic White Elders from Massachusetts Have Different Patterns of Carotenoid Intake and Plasma Concentrations. Journal of Nutrition, 135, pp. 1496-1502.
- Biacs, A.P., Daood, G.H., & Pause, A. (1989). Studies on the carotenoids pigments of paprika. Journal of agricultural and food chemistry., 37, 350-353.
CrossRef - Bohm, F., and Bitsch, R. (1999). Intestinal absorption of lycopene from different matrices and interactions to other carotenoids to the lipid status and antioxidant capacity of human plasma. European Journal of Nutrition, 38: 118-125.
CrossRef - Boileau, W.M., Moore, C., and Erdman, W.J. (1999). Carotenoid and Vitamin A. In: Antioxidants in human health and disease. Basu, K.T., Temple, J.N., and Gerg, L.M. CABI Publishing, New York.
- Bowen, Ε.P., Sapuntzakis, M.S., and Navsariwala, V.D. (2015). Carotenoids in Human Nutrition. Pigments in Fruits and Vegetables, 31-67.
CrossRef - Britton G. (1995). UV/visible spectroscopy. In Britton G, Liaaen-Jensen S, Pfander H (eds), Carotenoids: Spectroscopy, vol 1B, pp 13-63.
- Brubacher, G., Muller-Mulot, W., & Southgate, D.A. (1985). Methods for the determination of vitamins in foods. Elsevier Applied Science, London.
CrossRef - Bushway, R.J. (1985). Separation of carotenoids in fruits and vegetables by HPLC. Journal of Liquid Chromatography, 8,1527-1547.
CrossRef - Cabrera, P.K, Huo, T., Schwartz, J.S., and Failla, L.M. (2010) Digestive Stability and Transport of Norbixin, a 24-Carbon Carotenoid, across Monolayers of Caco-2 Cells. Journal of agricultural and food chemistry, 12; 58(9).
- Chisté, C.R., Yamashita, F., Gozzo, C.F., and Mercadante, Z.A (2011). Simultaneous extraction and analysis by high performance liquid chromatography coupled to diode array and mass spectrometric detectors of bixin and phenolic compounds from annatto seeds. Journal of Chromatography, 1218, pp. 57–63.
CrossRef - Daood, G.H., Vinkler, M., Markus, F., & Biacs, R.A. (1996). Antioxidant vitamin activities of spice red pepper (paprika) as affected by technological and varietal factors. Food Chemistry, 55, 365-372.
CrossRef - Deli, J., Matus, R., & Stochols, J. (1992). Carotenoid composition in the fruits of black paprika. J.Agric.Food.Chem., 40, 2072-2076.
CrossRef - Deli, J., and Molnar, P. (2002). Paprika Carotenoids: Analysis, Isolation, Structure, Elucidation. Current Organic Chemistry, 6 (13), 1197-1219.
CrossRef - Dimakou, C and Oreopoulou,V. (2012). Antioxidant activity of carotenoids against the oxidative destabilization of sunflower oil-in-water emulsions. LWT – Food Science and Technology, 46, pp. 393-402.
CrossRef - Faure. H., Galabert, G., Le Moel, G., and Nabet, F. (1999). Carotenoids: metabolism and physiology. Annales de Biologie Clinique., 57, 169-183.
- Gann, H.P., Deaton, R., Enk, E., van Breemen, B.R., Han, M., Lu, Y., and Ananthanarayanan, V. (2012). A Phase II randomized trial of lycopene-rich tomato extract among men with high-grade prostatic intraepithelial neoplasia (HGPIN). Cancer Res, 72; 3564
CrossRef
- Giuffrida, D., Dugo, P., Torre, G., Bignardi, C., Cavazza, A., Corradini, C., and Dugo, G. (2013). Characterization of 12 Capsicum varieties by evaluation of their carotenoid profile and pungency determination. Food Chemistry, 140, 794-802.
- Hadden WL, Watkins RH, Levy LW, regalado E, Rivadeneira DM, Van Breemen R, Schwartz SJ. (1999). Carotenoid composition of marigold (Tagetes erecta) flower extract used as nutritional supplement. Journal of agricultural and food chemistry, 47:4189-4194.
- Hakal, H.S. & Heinonen, M. (1994). Chromatography of purified lycopene. Journal of agricultural and food chemistry, 44, 1314-1316.
CrossRef - Handelman, G.J., Shen, B., & Krinsky, N.I. (1992). High resolution analysis of carotenoids in hunan plasma by HPLC. Methods in Enzymology, 213, 336-346.
CrossRef - Hart, J.D., & Scott, K., (1995). Development and evaluation of HPLC methods for the analysis of carotenoids in foods and the measurement of carotenoid content of vegetables commonly consumed in UK. Food Chemistry, 54, 101-111.
CrossRef - Khachik, F., Beecher, G.R., and Smith, J.C. (1997). Lutein, lycopene, and their oxidative metabolites in chemoprevention of cancer. J.Cell.Biochem. 22: 236-246.
- Kiokias, S. (2002). In vitro and in vivo antioxidant properties of natural carotenoid mixtures. Faculty of Life Sciences, School of Food Biosciences, The University of Reading.
- Kiokias, S. & Gordon, M. (2003a). Antioxidant properties of annatto carotenoids. Food Chemistry, 83, 523-529.
CrossRef - Kiokias, S and Gordon, M. (2003b). Dietary supplementation with a natural carotenoid mixture decreases oxidative stress. Europ. J. Clin. Nutr. 57: 1135-1140.
CrossRef - Kiokias, S. and Gordon M. (2004). Properties of carotenoids in vitro and in vivo. Food Reviews International, 20: 99-121.
CrossRef - Kiokias, S., and Oreopoulou, V. (2006). Antioxidant properties of natural carotenoid preparations against the AAPH-oxidation of food emulsions. Innovative Food Science & Emerging Technologies, 7, 132-139.
CrossRef - Kiokias, S., Varzakas, T., and Oreopoulou,V. (2008). In vitro activity of vitamins, flavonoids, and natural phenolic antioxidants against the oxidative deterioration of oil-based systems. Critical Reviews in Food Science and Nutrition, 48, 78–93.
CrossRef - Kiokias, S., Dimakou, C., and Oreopoulou, V. (2009a). Effect of natural carotenoid preparations against the autoxidative deterioration of sunflower oil-in-water emulsions. Food Chemistry, 114, 1278-1284.
CrossRef - Kiokias, S., Dimakou, C., and Oreopoulou,V. (2009b). In vitro antioxidant activity of synthetic beta- and carotene and natural carotenoid extracts against the oxidatative degradation of food emulsions. In beta-carotene, Dietary sources, Cancer and Cognition, (chapter-5) p.231-262. Nova Science.
- Kiokias, S., & Varzakas, T. (2014). Activity of flavonoids and beta-carotene during the auto-oxidative deterioration of model food oil-in water emulsions. Food Chemistry, 150C, 280-286.
CrossRef - Kovary, K., Lourain, T., Silva, C., Albano, F., Pires, M.B., Lage, S.L., and Felzenswalb, I. (2001).Biochemical behaviour of norbixin during in vitro DNA damage induced by reactive oxygen species. British Journal of Nutrition. 85: 431-440.
CrossRef - Lietz, G., & Henry, J.K. (1997). A modified method to minimize losses of carotenoids and tocopherols during HPLC analysis of red palm oil. Food Chemistry, 60, 109-117.
CrossRef - Liu., G., Gerken,H., Huang, J. and Chen, F. (2013) Engineering of an endogenous phytoene desaturase gene as a dominant selectable marker for Chlamydomonas reinhardtii transformation and enhanced biosynthesis of carotenoids. Plant Science, 208, pp 58-63.
CrossRef - Miki, W., Park, H.M., & Takeda, R. (1990). Separation of carotenoid esters using microwave oven. Paper presented on 9th International Symposium of Carotenoids, Japan.
- Pantavos, A., Ruite, R., Feskens, F.E., de Keyser, E.C., Hofman, A., Stricker, H.B., Franco, H.O., and Kiefte-de Jong, C.J. (2015). Total dietary antioxidant capacity, individual antioxidant intake and breast cancer risk: The Rotterdam study. International Journal of Cancer, 136, pp. 2178-2186.
CrossRef - Park, S-Y., Choi, S-R., Lim, S-H, Yeo,Y., Kweon, S-J., Bae, Y-S., Kim, K-W., Im, K-H., Ahn, S-K. (2014). Identification and quantification of carotenoids in paprika fruits and cabbage, kale, and lettuce leaves. Journal of the Korean Society for Applied Biological Chemistry, 57(3), pp 355-358.
CrossRef - Pérez-Gálvez A., and Mínguez-Mosquera M.I. (2004). Degradation, under non-oxygen-mediated autooxidation, of carotenoid profile present in paprika oleoresins with lipid substrates of different fatty acid composition, Journal of agricultural and food chemistry, 52 (2004) 632–637.
CrossRef - Rodriguez C, M., and Stange, C. (2013). Biosynthesis of carotenoids in carrot: An underground story comes to light. Archives of Biochemistry and Biophysics, 539, pp. 110-116.
CrossRef - Rodriguez-Amaya, D.B (2015). Status of carotenoid analytical methods and in vitro assays for the assessment of food quality and health effects. Current Opinion in Food Science, 1, pp 56–63.
CrossRef - Saleh, H.M., & Tan, B. (1991). Separation and identification of cis/trans carotenoid isomers. Journal of agricultural and food chemistry, 39, 1438-1443.
CrossRef - Scott, E.C and Eldridge, L.. (2005). Comparison of carotenoids content in fresh, frozen and canned corn. Journal of Food composition and Analysis, 18 551-559.
CrossRef - Scotter, M.J. (1995). Characterisation of the coloured thermal degradation products from annatto, and a revised mechanism for their formation. Food Chemistry, 53, 177-185.
CrossRef - Scotter, M.J., Wilson, L.A., and Appleton, G.P. (1998). Analysis of annatto food color formulation. Determination of colour components and thermal degradation products by HPLC. Journal of agricultural and food chemistry, 46, 1301-1308.
CrossRef - Scotter, M.J. (2009). The chemistry and analysis of annato food colouring; a review. Food Additives and Contaminants, 26(8), 1123-1145.
CrossRef - Shoefs, B., Bertram, M., and Lemoine, Y. (1995). Separation of photosynthetic pigments and their precursors by reversed phase HPLC using photodiode array detectors. Journal of Chromatography, 692, 239-245.
CrossRef - Shumskaya, Μ., and Wurtzel, E.T. (2013). The carotenoid biosynthetic pathway: Thinking in all dimensions. Plant Science, 208, pp 58-63.
CrossRef - Silva, F.A., Marcelo M.R., and Silva, M.C. (2013). Supercritical solvent selection (CO2versus ethane) and optimization of operating conditions of the extraction of lycopene from tomato residues: Innovative analysis of extraction curves by a response surface methodology and cost of manufacturing hybrid approach. The Journal of Supercritical Fluids, 95, pp 618–627.
CrossRef - Šivel, M., Krackmar, S., Fisera, M., Klejdus, B., and Kuban, V. (2014). Lutein Content in Marigold Flower (Tagetes erecta L.). Concentrates used for Production of Food Supplements. Czech J. Food Sci., Vol. 32, 2014, No. 6: 521–525.
- Topuz, A., Dincer, C., Özdemir, S.K., Feng, H., Kushad, M. (2011). Influence of different drying methods on carotenoids and capsaicinoids of paprika (Cv., Jalapeno). Food Chemistry, pp. 860–865.
CrossRef - Vargas. D.F., and and López, P.O. (1996). Correlation of HPLC and AOAC methods to assess all-trans lutein content in marigold flowers. J.Sci.Food.Agric., 72, 283-290.
CrossRef - Vargas, D.F., and López, P.O. (1997). Effects of enzymatic treatments on carotenoid extraction from marigold flowers (Tagetes erecta). Food Chemistry, 58 (3): 255-258.
CrossRef - Viuda-Martos, M., Ciro-Gomez, L.G., Ruiz-Navajas,Y., Zapata-Montoya, E.Z., Sendra, E., José A. Pérez-Álvarez, A.J. and Fernández-López. J. (2012). In vitro Antioxidant and Antibacterial Activities of Extracts from Annatto (Bixa orellana L.) Leaves and Seeds. Journal of Food Safety, 32, pp 399-406.
CrossRef - Xiao-Dan, F., Hou,Y., Huang, X, Qiu, T., Jiang JG (2015). “Ultrasound Enhanced Subcritical CO2 Extraction of Lutein from Chlorella pyrenoidosa.“Journal of agricultural and food chemistry, 63 (18), 4597-4605.
CrossRef - Zakaria, M., Simpson, K., & Brow, R.P. (1989). Use of R.P HPLC Analysis for the determination of provitamins A carotenoids in tomatoes. Journal of Chromatography, 176,109-117.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.








