Diagnostic Prediction of Progressive Atherosclerosis in High-Fat Meal Consumers and Their Common Pathological Mechanisms
 , Shan Li3, , 5and Jiuchen Wang1*
, Shan Li3, , 5and Jiuchen Wang1* 1Department of Reproductive Medical Center, Taihe Hospital, Hubei University of Medicine, Shiyan, China
2Oil Crops Research Institute, Chinese Academy of Agricultural Sciences, Wuhan, China
3Department of Pulmonary and Critical Care Medicine, Sinopharm Dongfeng General Hospital, Hubei University of Medicine, Shiyan, China
4Department of Cardiology, Zhongnan Hospital, Wuhan University, Wuhan, China
5Institute of Myocardial Injury and Repair, Wuhan University, Wuhan, China
Corresponding Author’s Email: jiuchenwang@tmu.edu.cn
DOI : http://dx.doi.org/10.12944/CRNFSJ.13.3.26
Download this article as:
![]()
The occurrence of high-fat meals among the general population is on the rise, and ample evidence indicates their involvement in the progression of atherosclerosis (AS). Nevertheless, it is imperative to investigate vital research inquiries concerning the shared pathological processes, associations, and means of foretelling the susceptibility to AS in individuals who consume meals potentially rich in fat. We obtained high-fat meal (HFM) and progressive coronary artery disease (PCAD) gene expression profiles from the database of gene expression. Following the identification of the genes with differential expression common to HFM and PCAD, we performed a series of bioinformatics analyses, including machine learning and immune infiltration, to further our understanding of this issue. Finally, quantitative real-time polymerase chain reaction (qRT-PCR) validated the critical genes in murine serum. Using a combination of 20 widely used machine learning algorithms, we successfully created a reliable diagnostic predictor to evaluate the vulnerability for PCAD development in a particular HFM. Additionally, an in-depth exploration of their mutual connections within domains such as immunology enabled us to recognize possible mechanisms underlying the co-onset of HFM and PCAD and find the critical genes in the progressive AS model group. In this investigation, we employed machine learning to anticipate the pathogenesis and examine the mechanisms of two correlated conditions: HFM and PCAD. Our discoveries present innovative perspectives on the mechanistic comprehension of HFM and PCAD, which may prove beneficial for advancing forthcoming preemptive, individualized, and precise medical strategies.
KEYWORDS:Diagnostic biomarkers; Differentially expressed genes (DEGs); High-fat meal (HFM); Machine-learning; Progressive atherosclerosis;
Introduction
The prevalence of high-fat meals (HFM) is rising due to shifting traditional dietary habits, leading to many health issues. The Global Burden of Disease study indicates that cases of ischemic heart disease globally rose from over 100 million in 1990 to more than 180 million in 2019. The reduction in the incidence of ischemic heart disease in certain areas of the USA and UK, once ascribed to the management of risk factors, has either decelerated or ceased between 2014 and 2019. Atherosclerotic cardiovascular disease has emerged as a global concern, and even high-income countries may be losing ground in terms of prevention.1,2 AS was previously regarded as an unavoidable process of progressive degradation over time. Current research, however, indicates a more dynamic and discontinuous evolution of atheromata, with the possibility of full retreat, which further highlights the importance of the relevant research.2-6
Moreover, some research indicates that the ingestion of a high-fat meal may facilitate the onset of AS, and it may function independently of the negative effects of cholesterol.7,8 Given the potential risk HFM poses for developing AS, comprehending the underlying mechanism is of utmost importance. Hence, this research aimed to investigate the common pathogenesis of HFM and AS. As we know, the molecular mechanism is an important part that affects and analyzes the progression of AS.9 We acquired gene expression profiles from the Gene Expression Omnibus (GEO) for this purpose. We found differentially expressed genes (DEGs) that overlap in both HFM and progressive coronary artery disease (PCAD). Subsequently, bioinformatics analyses were performed to summarize the findings visually and enhance the current understanding of this topic.
Additionally, an accurate predictor for diagnosing PCAD, specifically designed for individuals with HFM, was developed. By establishing a comprehensive causal connection between these two diseases based on their shared pathogenesis, the outcomes of this study can provide valuable insights for subsequent studies and clinical implementation regarding HFM and PCAD.
Materials and Methods
Data Acquisition, Preparation, and Statistical Administration
GEO (http://www.ncbi.nlm.nih.gov/geo/) is a comprehensive database that has collected and organized the high-throughput genomic data (including microarray chips, second-generation sequencing, and other forms) uploaded by researchers worldwide, which houses gene expression information about diverse diseases and is freely accessible to the public. The specific dataset analyzed in this study was derived from the GEO database. The “normalize between arrays” function from the R package “limma” was utilized to standardize and regulate the HFM and PCAD datasets.10 The aforementioned analyses were performed utilizing various R software packages and the integrated Python module ‘sklearn’.11 Unless explicitly mentioned, the analytical procedure employed was the Wilcoxon rank-sum test. Asterisks (*, **, ***) may appear in certain Figures, indicating p-values of less than 0.05, 0.01, and 0.001, respectively.
Identification of Shared Differentially Expressed Genes Between High-Fat Meals and Progressive Coronary Artery Disease
For the current investigation, we analyzed differential expression utilizing the ‘limma’ package in R.10 To ensure precision, we employed a screening method for DEGs with a criterion of a p-value < 0.05 and a Log2 |fold change| > 0.95. By conducting the DEG screening on both HFM and PCAD datasets, we compared the outcomes to identify the common DEGs in both datasets.
Functional Enrichment Analysis
We utilized the R package ‘clusterprofiler’ to analyze functional enrichment, assessing Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Our investigation concentrated on identifying common DEGs.12 The identification of the most enriched GO terms and KEGG pathways was predicated on a significantly adjusted p-value of less than 0.05, which constituted the threshold for significance.
Predicting Potential Drugs Based on Gene Interaction Networks
We developed a network to illustrate the interactions of human genes for the purpose of annotating drug-target and disease-associated genes. We computed the most direct route between the medication target genes and disease-associated genes to examine the network architecture. To quantify the proximity between the drug and the disease, we obtained a z-score through a z-test. We performed random perturbation by repeating the sampling N times to establish the background distribution. Then, we calculated the corresponding p-value for the z-score and adjusted it for multiple tests. By analyzing statistically significant results, we identified potential drugs that can be treatments for the disease.13 Furthermore, we employed the STRING website to develop protein-protein interaction (PPI) networks, enhancing our comprehension of the relationships among primary targets.
Machine Learning
The identification of critical genes is vital in constructing the diagnostic predictor. This study utilized the ‘RFE’ technique to ascertain the optimal number of genes for modeling purposes. A synthesis of the linear algorithm ‘LASSO’ and the non-linear technique ‘Random Forest’ was employed to enhance the identification of potentially pertinent genes. The finalized selection of feature genes was subsequently used in creating the formal model, ultimately serving as the dependable PCAD diagnostic predictor for patients affected by HFM.
The complete AMI merged dataset was subjected to randomization to conduct a formal modeling analysis. It was subsequently partitioned into a training set and a test set with a ratio of 7.5:2.5. As per the ‘no-free-lunch’ theorem, the superiority of a particular machine learning algorithm on one assessment measurement might be accompanied by a compromise in other measurements.14 Put differently, no algorithm can be deemed flawless. Nevertheless, by exhaustively exploring various mainstream machine learning algorithms and evaluating their performance, we managed to identify the most optimal one overall. For this investigation, we conducted a comparative analysis of 20 machine learning algorithms, including Linear Regression, Ridge Regression, RidgeCV, Linear LASSO, LASSO, ElasticNet, BayesianRidge, Logistic Regression, SGD, SVM, KNN, Naive Bayes, Decision Tree, Bagging, Random Forest, Extra Tree, AdaBoost, GradientBoosting, Voting, and ANN.
Decision Curve Analysis (DCA)
Clinical models are often evaluated using ROC-AUC, accuracy, precision, recall, and F1-score, omitting clinical outcomes. However, DCA compares the clinical benefits obtained using different diagnostic predictors. The superiority of the curve’s localization indicates a better prediction from a clinical perspective.
Clinical models are generally assessed using metrics like ROC-AUC, Accuracy, Precision, Recall, and F1-score, usually overlooking clinical outcomes. Nonetheless, DCA allows for comparing clinical advantages achieved through the utilization of diverse diagnostic predictors.15,16 The superior localization of the curve reflects improved clinical prediction.
Analysis of the Immune Microenvironment
To detect duplicated content, researchers employed CIBERSORT2, a device employed to evaluate the existence of diverse invading immune cells.17,18 In total, 22 different immune cell types were quantified. Pearson’s technique was utilized to examine the correlation between immune cell types and GLS and SLC31A1. The results were visualized using the R tool ‘ggplot2.’
Animals and Procedures
This study utilized male ApoE−/− mice at an age of 6 weeks. The mice were maintained in typical pathogen-free settings. In this experiment, the mice were randomly allocated to two groups: the model group (n = 6), which received a high-fat diet, and the control group (n = 6), which was provided with a conventional diet. Following this, blood and aorta samples were obtained from each mouse in the seventh week after the initiation of their respective diets.
Results
Identification of Common DEGs
 |
Figure 1: The detection of shared DEGs in HFM and PCAD datasets was conducted. (A, B) The analysis involved the visualization of upregulated and downregulated DEGs separately using volcano plots in the HFM and PCAD datasets. |
Our examination of the HFM dataset revealed 667 genes exhibiting differential expression. Among these genes, 289 were upregulated, while 378 were downregulated (Fig. 1A). Similarly, in the PCAD dataset, we observed 1025 genes that exhibited differential expression. Out of these genes, 430 were found to be upregulated, whereas 595 were downregulated (Fig. 1B). By comparing the DEGs from both datasets, we uncovered a set of 21 genes that were commonly differentially expressed (Fig. 1C).
Functional Enrichment Analysis, Correlation, and PPI Network Analysis
 |
Figure 2: Functional annotation for shared differentially expressed genes (DEGs). (A) Dot plot illustrating the results of GO enrichment analyses for shared DEGs. (B) Dot plot showcasing the findings of KEGG enrichment analyses for shared DEGs. |
We conducted an analysis of GO and KEGG enrichments to investigate gene function. We conducted a conventional functional enrichment analysis and discovered 15 statistically significant GO keywords and 23 KEGG pathways. The top 15 enrichments can be observed in Fig. 2 A and 2B. The enriched GO terms are shown in Fig. 2C. In terms of GO enrichment, the common DEGs mainly focused on protein heterodimerization activity. The KEGG analysis revealed that the primary pathways of the common DEGs include the Tumor necrosis factor (TNF) signaling pathway (Fig. 2D), Hepatitis B, Human cytomegalovirus infection, Human papillomavirus infection, and the PI3K-Akt signaling pathway. TNF is a pro-inflammatory cytokine crucial for mammalian immunity and the maintenance of cellular homeostasis. Dysregulation of TNF receptor (TNFR) signaling correlates with multiple inflammatory disorders, including diverse forms of arthritis and inflammatory bowel disease. TNF conveys signals through TNFR1 and TNFR2, leading to the activation of nuclear factor-κB (NF-κB) and promoting cell survival. A significant feature of TNF-dependent NF-κB signaling is the intricate regulation of receptor-interacting serine/threonine protein kinase 1 (RIPK1) activation through both linear and branched ubiquitylation.19 To visualize the PPI network diagram, the frequent DEGs were input into the STRING database. Fig. 2B demonstrates that central positions in the network diagram are occupied by genes like TIMP3 and SEC16B.
Pre-Modeling: A Comprehensive Methodology for Gene Feature Selection
 |
Figure 3: Integrative strategy for selecting genes for features. (A) A scree plot demonstrates the fluctuation in RMSE values as the quantity of variations (i.e., feature genes) employed in formal modeling varies. |
To find the appropriate genes for formal modeling, we utilized a mixed strategy incorporating both linear (LASSO) and non-linear (Random Forest) methodologies. We initially established that the optimal number of genes for the RFE method is five. Subsequently, we observed that the RMSE value displayed minimal fluctuations, indicating consistent predictive power (Fig. 3A). The importance ranking of the twenty most significant genes identified by the Random Forest method revealed SFTPC, ARHGEF35, PDZD7, NPBWR1, and TIMP3 as the most crucial (Fig. 3C). By utilizing the LASSO method, we minimized the binormal deviance (Fig. 3B) and assigned coefficients to each gene (Fig. 3D). Moreover, we managed to identify the top 6 genes with the highest weights (Fig. 3E). Ultimately, for formal modeling, we selected the genes that were identified as overlapping by both the Random Forest algorithm and the LASSO approach.
Formal Modeling: Creating a Diagnostic Prediction for HFM in PCAD Patients
 |
Figure 4: Multifaceted assessment of 20 prevalent predictors for machine-learning diagnostics. (A) The visualization plot illustrates the metrics of accuracy, recall, and F1-score for both the training and testing datasets. |
This experiment evaluated the efficacy of 20 various algorithms in their capacity to reliably anticipate outcomes and attributes. Among these methods, the Artificial Neural Network exhibited the maximum efficacy in Accuracy, Precision, Recall, and F1-score (Fig. 4A), closely succeeded by Logistic Regression and subsequently by Stochastic Gradient Descent. Moreover, the ANN and Logistic Regression algorithm also obtained the highest ROC-AUC value of 0.867 when applied to the test dataset (Fig. 4B). It is essential to highlight the fact that dyslipidemia, a well-established indicator for PCAD, demonstrated an equivalent ROC-AUC value of 0.867. This implies that the ANN and Logistic Regression algorithm can yield a comparable impact (Fig. 4C).
Investigation of the Implications of Feature Genes in Immunology
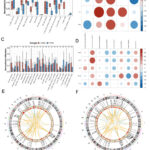 |
Figure 5: The immunological implications of feature genes were investigated in this study. (A) The study compared the infiltrating immune cells in the HFM and control groups. (B) A correlation graph was generated to examine the association between prevalent genes and immune cells. |
An analysis utilizing the Cibersort platform revealed a significant disparity in T_cells_CD8 expression levels between the HFM samples and the control group (Fig. 5A). Furthermore, the investigation of PCAD displayed a statistical disparity in Dendritic_cells_resting when compared to the control group (Fig. 5C), and no shared differential regulations of immune cells were observed. Notably, the prevalent DEGs SLC8A3, SOX18, NAT8L, and TAS2R4 were identified as being linked to memory B cells. CIBERSORT, T_cells_CD8_CIBERSORT, T_cells_CD4_naive_CIBERSORT, activated NK cells CIBERSORT, and Neutrophils CIBERSORT (Fig. 5B). The common DEGs TAS2R4, SOX18, TIMP3, SEC16B, and NAT8L were associated with T_cells_CD4_memory_resting_CIBERSORT, T_cells_follicular_helper_CIBERSORT, Dendritic_cells_resting_CIBERSORT, Eosinophils_CIBERSORT, Neutrophils_CIBERSORT, P.value_CIBERSORT, Correlation_CIBERSORT, and RMSE_CIBERSORT (Fig. 5D). The coexistence of SOX18, NAT8L, and TAS2R4, alongside Neutrophils_CIBERSORT in immune cells, displays a strong correlation within both HFM and PCAD. The visualization of circus plots effectively showcases the chromosomal location of these genes in both HFM and PCAD. The outer ring represents the segmented genome, while the inner ring exhibits the logFC associated with each gene, with the red line denoting up-regulation and the blue line indicating down-regulation (Fig. 5 E and 5F).11
Validation of Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)–Based Machine Learning Analysis
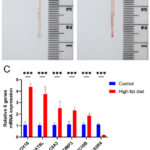 |
Figure 6: Analysis of atherosclerotic Lesions and serum qRT-PCR at the seventh week. (A) Oil Red O staining of atherosclerotic lesions in the aorta of the control group. (B) Oil red O staining of atherosclerotic lesions in the aorta of the high-fat diet group. |
To validate the results of the machine learning analysis, SOX18, NAT8L, SLC8A3, TIMP3, SEC16B, and TAS2R4 were selected for qRT-PCR. The expression levels of SOX18, NAT8L, SLC8A3, TIMP3, and SEC16B were consistently increased, while TAS2R4 expression levels were reduced in the high-fat diet group (Fig. 6).
Discussion
AS, the predominant form of cardiovascular disease, is a major factor in morbidity and mortality globally.20,21 Substantial data indicates that dietary components directly influence the progression of AS and can alter traditional risk variables, including plasma lipids, blood pressure, and plasma glucose levels.22 High-fat meals display substantial detrimental effects on cardiovascular well-being, resulting in inflammation after a meal, harmful impacts on the brain and cognitive abilities, and contributing to global mortality.23-26 Thus, it is crucial to investigate the mechanisms underlying the interaction between AS and high-fat meals while exploring potential targets for the treatment of these conditions. Our research has successfully identified drug targets for high-fat meals and the associated cardiovascular complications, which establishes a solid foundation for the diagnosis and management of associated disorders.
The data of GEO is characterized by its richness and international openness, and is widely used in mechanism analysis research.27,28 The GEO database provided the gene expression data for PCAD and HFM. Through analyzing this data, we identified 21 genes that showed DEGs. Thereafter, we performed a series of systematic and bioinformatics analysis. Additionally, we developed a reliable predictor by employing 20 well-established machine-learning algorithms to evaluate the probability of PCAD occurrence in HFM patients. By conducting these procedures, we were able to unveil the fundamental mechanisms implicated in the co-pathogenesis of HFM and PCAD.
In both the LASSO and Random Forest algorithms, the most significant genes identified among the DEGs were SOX18, NAT8L, SLC8A3, TIMP3, SEC16B, and TAS2R4. The findings indicate that SOX18 is implicated in the proliferation of vascular cells and may contribute to the pathogenesis of AS.29 SOX18, as a potential target for anti-angiogenic therapies in human cancer, contributes to the proliferation of vascular cells and indicates that this transcription factor may be implicated in AS.30,31 NAT8L is involved in autophagy by influencing mitochondrial function, and NUPR1 and TIA1 may control the levels of TGFB2-OT1 and TGFB2-OT1. TGFB2-OT1 targets CERS1, NAT8L, and LARP1 MIR3960, MIR4488, and MIR4459, which target essential proteins implicated in autophagy and inflammation, respectively.32 Cacna1d, Serpine1, Slc8a3, Trpc4, Myoz2, and Slc25a23 may participate in the control of calcium metabolic pathways and are pivotal in hypertension. The differentially expressed genes associated with calcium metabolism may serve as prognostic indicators for hypertension.33 TIMP1 overexpression or TIMP3 silencing has consistently been linked to cancer development and unfavorable patient prognosis among the four TIMPs. Future initiatives will utilize TIMP in liquid biopsy specimens as a biomarker for cancer prognosis. TIMPs may serve as biomarkers in the bodily fluids of patients.34 TIMP-1 and MMP-9 exhibit promising capabilities as reliable biomarkers for early prognosis in patients in the intensive care unit (ICU).35 TIMP3 and RECK have been shown to enhance the inhibitory effects of interleukin-32a (IL-32a) on endothelial inflammation, smooth muscle cell activation, and AS progression.36 The immunological and inflammatory emphasis of HFM and PCAD has been verified through both DEGS and immunoassays. Our findings receive partial support from the identification of the ‘TNF signaling pathway’ in the KEGG outcomes. In the contemporary age of intricate food patterns, we can utilize this pathway as a first measure to investigate the essential mechanisms of damage in the circulatory system induced by HFM, while revealing novel preventive strategies. SEC16B is a significant biological candidate gene for obesity-related disorders, as it encodes both the long Sec16L and the short Sec16 proteins, which are crucial for the vesicular transport of secretory substances from the endoplasmic reticulum (ER) to the Golgi apparatus.37,38 We found that the SEC16B/RASAL2 SNP showed a substantial master effect on BMI (p=0.001), demonstrating the importance of specific environmental genetic effects and contributing to the growing literature highlighting the possible importance of physical activity in mitigating potential genetic risk.39 Reports indicate that TAS2R4 has sensitivity to cholesterol in its signaling activity. Research has investigated the correlations among 15 variants in two TASR genes and 12 AR genes, confirming genetic links with specific obesity-related traits discovered in earlier studies.40 The effect on the respective phenotype for six mutations was swiftly correlated with the gene’s known function. These six variants are located inside four AR genes (SIM1, FOS, MCHR2, and LEPR) and two TASR genes (TAS2R4 and TAS2R9). This study provides new insights into the functional importance of the eight remaining variants in the AR gene (MTNR1B, ALDH2, GPR179, GPRC5C, ALDH3B2, GRM1, ALDH1B1, and P2RX7) by examining their specific associations with certain obesity-related symptoms. The majority of the 15 statistically significant correlations were age-specific, suggesting that adolescents and older adults regulate fat deposition, weight gain, and metabolism differently.41 These crucial DEGs provide insights into the clinical features of PCAD resulting from HFM, suggesting that HFM may induce thrombosis and the creation of PCAD through vascular inflammation.
Based on our extensive investigation that utilized 20 mainstream algorithms for machine learning, it has come to our attention that the ANN and logistic regression display the highest level of predictive performance.14,42 In order to confirm the credibility of our discoveries, we proceeded to compare the effectiveness of ANN and logistic regression with the biomarker dyslipidemia, which is widely recognized for its connection to AS. To our astonishment, we discovered that the predictive ability of ANN (Auc=0.867) and logistic regression was equal to that of dyslipidemia (Auc=0.867). This observation suggests that the ANN and logistic regression algorithm predictor demonstrates remarkable precision when it comes to diagnosing AS in individuals afflicted with HFM. Additionally, it presents an alternative choice to traditional prediction methods and encourages further investigation into prospective novel biomarkers that offer promising capabilities.
Both immunoassays and DEGs have confirmed the focus on immune and inflammatory aspects connected to HFM and PCAD. Our research findings are partially substantiated by the discovery of the ‘TNF signaling pathway’ in the results obtained from KEGG. Given the intricate nature of dietary habits at present, this pathway can serve as an initial investigative point for understanding the fundamental mechanisms of circulatory system damage caused by HFM. Additionally, it offers an opportunity to explore innovative preventive measures.
Recruiting participants for this study posed a limitation due to the extended cyclic progression of AS. Nevertheless, as an innovative inquiry, we employed cutting-edge methodologies like machine learning and network pharmacology to investigate the correlation between HFM and PCAD. Our discoveries offer novel perspectives into the mechanisms underlying HFM and PCAD, bearing significant implications for forthcoming research in preventive, personalized, and precision medicine.
Conclusion
We obtained the gene expression profiles of HFM and PCAD from the gene expression database. After identifying the differentially expressed genes shared by HFM and PCAD, we conducted a series of bioinformatics analyses, including machine learning and immune infiltration, to further understand this issue. Finally, the essential genes in the mouse serum were determined by qRT-PCR. Combining 20 widely used machine learning algorithms, we successfully created a reliable diagnostic predictor to assess the vulnerability of PCAD development in a specific HFM and compared it with the results of dyslipidemia. Furthermore, an in-depth exploration of their interrelationships in fields such as immunology enables us to identify the possible mechanisms of the co-onset of HFM and PCAD, and to identify key genes in the progressive AS model group. Our findings provide a theoretical basis and technical guidance for the prevention of PCAD from the perspective of HFM. Undoubtedly, it is crucial to conduct larger-scale and deeper clinical trials in the future.
Acknowledgement
We like to convey our profound appreciation to public databases, such as GEO and STRING, for offering accessible and high-quality research resources.
Funding Sources
This study was supported by the National Natural Science Foundation of China (82200974).
Conflict of Interest
The authors do not have any conflicts of interest.
Data Availability Statement
The article incorporates the study’s original contributions. Further inquiries may be directed to the respective writers.
Ethics Statement
The study employed published research and consortia that provide publicly available aggregate data. All original investigations had approval from the appropriate ethical review board, and participants provided informed consent. Moreover, the study did not utilize individual-level data. Consequently, the endorsement of a new ethical review commission is unnecessary. All animal procedures adhered to the Animal Care and Use Guidelines and received approval from the Animal Welfare Committee.
Informed Consent Statement
The analysis utilizes published studies and consortia that offer publicly accessible aggregated data. All original study participants provided informed consent.
Clinical Trial Registration
The research utilized published studies and consortia that offered publicly accessible statistics. The experiment was registered in a publicly available registry before patient enrollment and does not necessitate registration in a new registry.
Permission to Reproduce Materials from Other Sources
Not applicable.
Author Contributions
- Yaxi Xu: Conceptualization, Data Curation, Formal Analysis, Methodology, Visualization, Writing-Original Draft, Writing-Review & Editing.
- Shan Li: Writing-Review & Editing.
- Ze Chen: Funding acquisition, Supervision, Writing-Review & Editing.
- Jiuchen Wang: Supervision, Writing-Review & Editing.
References
- Mensah GA, Roth GA, Ademi Z, et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J. Am. Coll. Cardiol. 2020;
- Libby P. The changing landscape of atherosclerosis. Nature. 2021/04/01 2021;592(7855):524-533.
CrossRef - Kubo T, Maehara A, Mintz GS, et al. The Dynamic Nature of Coronary Artery Lesion Morphology Assessed by Serial Virtual Histology Intravascular Ultrasound Tissue Characterization. Am. Coll. Cardiol. 2010;55(15):1590-1597.
CrossRef - Deliargyris EN. Intravascular ultrasound virtual histology derived thin cap fibroatheroma now you see it, now you don’t. Am. Coll. Cardiol. 2010;55(15):1590-1597.
CrossRef - Wang X, Ge J. Atherosclerotic Plaque Healing. Engl. J. Med. 2021;384(3):293.
CrossRef - Mendieta G, Pocock S, Mass V, et al. Determinants of Progression and Regression of Subclinical Atherosclerosis Over 6 Years. J Am Coll Cardiol. 2023;82(22):2069-2083.
CrossRef - Vogel RA, Corretti MC, Plotnick GD. Effect of a Single High-Fat Meal on Endothelial Function in Healthy Subjects. Am. Coll. Cardiol. 1997;79(3):350-354.
CrossRef - Zimmerman B, Kundu P, Rooney WD, et al. The Effect of High Fat Diet on Cerebrovascular Health and Pathology: A Species Comparative Review. Molecules. 2021;26(11):3406.
CrossRef - ackson A-O, Regine MA, Subrata C, et al. Molecular mechanisms and genetic regulation in atherosclerosis. IJC Heart & Vasculature. 2018; 21:36-44.
CrossRef - Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47-e47.
CrossRef - Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: Machine Learning in Python. JMLRorg. 2011;(85)
- Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The innovation. 2021;2(3)
CrossRef - Guney E, Menche J, Vidal M, et al. article network-based in silico drug efficacy screening. 2017.
CrossRef - Wolpert DH, Macready WG. No free lunch theorems for optimization. IEEE Trans. Evol. Comput. 1997;1(1):67-82.
CrossRef - Vickers AJ, Elkin EB. Decision curve analysis: a novel method for evaluating prediction models. Decis. Making. 2006;26(6):565-574.
CrossRef - Vickers AJ, Calster BV, Steyerberg EW. A simple, step-by-step guide to interpreting decision curve analysis. Progn. Res. 2019;3.
CrossRef - Chen B, Khodadoust MS, Liu CL, et al. Profiling tumor infiltrating immune cells with CIBERSORT. Cancer Syst. Biol. Methods Protoc. 2018:243-259.
CrossRef - Craven KE, Gökmen-Polar Y, Badve SS. CIBERSORT analysis of TCGA and METABRIC identifies subgroups with better outcomes in triple negative breast cancer. Rep. 2021;11(1):4691.
CrossRef - Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Rev. Immunol. 2015;15(6):362-374.
CrossRef - Rafieian-Kopaei M, Setorki M, Doudi M, et al. Atherosclerosis: process, indicators, risk factors and new hopes. Int J Prev Med. Aug 2014;5(8):927-46.
- Björkegren JL, Lusis AJ. Atherosclerosis: recent developments. Cell. 2022.
CrossRef - Forouhi NG, Krauss RM, Taubes G, et al. Dietary fat and cardiometabolic health: evidence, controversies, and consensus for guidance. Bmj. 2018;361.
CrossRef - Cerf ME. High Fat Programming and Cardiovascular Disease. Medicina-Lithuania. 2018;54(5)86.
CrossRef - Freeman LR, Haley-Zitlin V, Rosenberger DS, et al. Damaging effects of a high-fat diet to the brain and cognition: A review of proposed mechanisms. Nutritional Neuroscience. Nov 2014;17(6):241-251.
CrossRef - Herieka M, Erridge C. High-fat meal induced postprandial inflammation. Nutr. Food Res. 2013.
CrossRef - Zimmerman B, Kundu P, Rooney WD, et al. The Effect of High Fat Diet on Cerebrovascular Health and Pathology: A Species Comparative Review. Molecules. 2021;26(11)3406.
CrossRef - Li LY, Wu JX. Analysis of hub genes and molecular mechanisms in non-alcoholic steatohepatitis based on the gene expression omnibus database. Zhonghua Yi Xue Za Zhi. 101(40):3317-3322.
- Clough E, Barrett T. The Gene Expression Omnibus Database. Methods Mol. Biol. Springer New York; 2016:93-110.
CrossRef - García-Ramírez M, Martínez-González J, Juan-Babot JO, et al. Transcription factor SOX18 is expressed in human coronary atherosclerotic lesions and regulates DNA synthesis and vascular cell growth. Arterioscler Thromb Vasc Biol. Nov 2005;25(11):2398-403.
CrossRef - Young N, Hahn CN, Poh A, et al. Effect of Disrupted SOX18 Transcription Factor Function on Tumor Growth, Vascularization, and Endothelial Development. Natl. Cancer Inst. 2006;98(15):1060-1067.
CrossRef - García-Ramírez M, Martínez-González J, Juan-Babot JO, et al. Transcription Factor SOX18 Is Expressed in Human Coronary Atherosclerotic Lesions and Regulates DNA Synthesis and Vascular Cell Growth. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25:2398-2403.
CrossRef - Huang S, Lu W, Ge D, et al. A new microRNA signal pathway regulated by long noncoding RNA TGFB2-OT1 in autophagy and inflammation of vascular endothelial cells. Autophagy. 2015;11(12):2172-2183.
CrossRef - Chang X, Guo L, Zou L, et al. Identification and Validation of Novel Biomarkers Related to the Calcium Metabolism Pathway in Hypertension Patients Based on Comprehensive Bioinformatics Methods. Health. 2024.
CrossRef - Jackson HW, Defamie V, Waterhouse P, et al. TIMPs: versatile extracellular regulators in cancer. Rev. Cancer. 2017;17(1):38-53.
CrossRef - Duda I, Krzych Ł, Jędrzejowska-Szypułka H, et al. Plasma matrix metalloproteinase-9 and tissue inhibitor of matrix metalloproteinase-1 as prognostic biomarkers in critically ill patients. Open Medicine. 2020;15(1):50-56.
CrossRef - Son DJ, Jung YY, Seo YS, et al. Interleukin-32α Inhibits Endothelial Inflammation, Vascular Smooth Muscle Cell Activation, and Atherosclerosis by Upregulating Timp3 and Reck through suppressing microRNA-205 Biogenesis. Theranostics. 2017;7(8):2186-2203.
CrossRef - Budnik A, Heesom KJ, Stephens DJ. Characterization of human Sec16B: indications of specialized, non-redundant functions. Rep. 2011;1(1):77.
CrossRef - Hotta K, Nakamura M, Nakamura T, et al. Association between obesity and polymorphisms in SEC16B, TMEM18, GNPDA2, BDNF, FAIM2 and MC4R in a Japanese population. J Hum Genet. 2009;54(12):727-31.
CrossRef - Young KL, Monda KL, Demerath EW, et al. Abstract P157: Does Physical Activity Modify the Association of 15 Well-established Obesity Loci with BMI: The ARIC Study. Am Heart Assoc; 2013.
CrossRef - Pydi SP, Jafurulla M, Wai L, et al. Cholesterol modulates bitter taste receptor function. Biochim Biophys Acta. 2016;1858(9):2081-2087.
CrossRef - Cirera S, Clop A, Jacobsen MJ, et al. A targeted genotyping approach enhances identification of variants in taste receptor and appetite/reward genes of potential functional importance for obesity-related porcine traits. Anim Genet. 2018;49(2):110-118.
CrossRef - Xie J, Luo X, Deng X, et al. Advances in artificial intelligence to predict cancer immunotherapy efficacy. Immunol. 2023; 13:1076883.
CrossRef
Accepted on: 22 Jul 2025
Second Review by: Eren Canbolat
Final Approval by: Dr. Aly El Sheikha





Web of Science Coverage
Emerging Sources Citation Index (ESCI)
2024 Journal Impact Factor: 1.1
Scopus Journal Metrics
CiteScore 2024: 1.9
CiteScore Details
Sustainable Nutrition: Food Systems, Nutrient Retention, and Public Health Impact
![]()
This journal is a member of, and subscribes to the principles of, the Committee on Publication Ethics (COPE)








